
Kembali otak-atik Ubuntu Linux.
Fokusnya adalah optimalisasi kemampuan ubuntu untuk pengajaran kimia dan ilmu
kimia sendiri. Dari list synatic package manager (SPM) ubuntu cukup banyak
aplikasi terkait kimia yang memungkinkan digunakan dalam mendukung pembelajaran
kimia di sekolah.
Beberapa deskripsi aplikasi-aplikasi tersebut sebagai berikut:
Beberapa deskripsi aplikasi-aplikasi tersebut sebagai berikut:
Xdrawchem (Chemical structures and
reactions editor)
Xdrawchem adalah editor 2D untuk struktur dan reaksi kimia. Kemampuannya merupakan bayangan program komersial ChemDraw, terutama untuk 2D-nya.
Xdrawchem adalah editor 2D untuk struktur dan reaksi kimia. Kemampuannya merupakan bayangan program komersial ChemDraw, terutama untuk 2D-nya.
GChempaint (2D chemical structures
editor for the GNOME2 desktop)
GChempait adalah aplikasi yg mirip dengan Xdrawchem, namun ditambah dengan kemampuannya membuat dokumen dengan berbagai interface. Gambar berbagai molekul hasil GChempaint ini dapat disearch pada NIST Webbook and PubChem.
GChempait adalah aplikasi yg mirip dengan Xdrawchem, namun ditambah dengan kemampuannya membuat dokumen dengan berbagai interface. Gambar berbagai molekul hasil GChempaint ini dapat disearch pada NIST Webbook and PubChem.
Chemtool (Chemical structures
drawing program)
Chemtool is a GTK+ based 2D chemical structure editor for X11. It supports many bond styles, most forms of text needed for chemical typesetting and splines/arcs/curved arrows.
Drawings can be exported to MOL and PDB format, SVG or XFig format for further annotation, as a PiCTeX drawing, as a bitmap or as Postscript files (several of these through XFig’s companion program transfig).
The package also contains a helper program, cht, to calculate sum formula and (exact) molecular weight from a chemtool drawing file. Cht can either be called directly by Chemtool or on the console.
Chemtool is a GTK+ based 2D chemical structure editor for X11. It supports many bond styles, most forms of text needed for chemical typesetting and splines/arcs/curved arrows.
Drawings can be exported to MOL and PDB format, SVG or XFig format for further annotation, as a PiCTeX drawing, as a bitmap or as Postscript files (several of these through XFig’s companion program transfig).
The package also contains a helper program, cht, to calculate sum formula and (exact) molecular weight from a chemtool drawing file. Cht can either be called directly by Chemtool or on the console.
Openbabel (Chemical toolbox
utilities)
Open Babel is a chemical toolbox designed to speak the many languages of chemical data. It allows to search, convert, analyze, or store data from molecular modeling, chemistry, solid-state materials, biochemistry, or related areas. Features include:
* Hydrogen addition and deletion
* Support for Molecular Mechanics
* Support for SMARTS molecular matching syntax
* Automatic feature perception (rings, bonds, hybridization, aromaticity)
* Flexible atom typer and perception of multiple bonds from atomic coordinates
* Gasteiger-Marsili partial charge calculation
Open Babel is a chemical toolbox designed to speak the many languages of chemical data. It allows to search, convert, analyze, or store data from molecular modeling, chemistry, solid-state materials, biochemistry, or related areas. Features include:
* Hydrogen addition and deletion
* Support for Molecular Mechanics
* Support for SMARTS molecular matching syntax
* Automatic feature perception (rings, bonds, hybridization, aromaticity)
* Flexible atom typer and perception of multiple bonds from atomic coordinates
* Gasteiger-Marsili partial charge calculation
File formats Open Babel supports
include PDB, XYZ, CIF, CML, SMILES, MDL, Molfile, ChemDraw, Gaussian, GAMESS,
MOPAC and MPQC.
This package includes the following
utilities:
* babel: Convert between various chemical file formats
* obenergy: Calculate the energy for a molecule
* obminimize: Optimize the geometry, minimize the energy for a molecule
* obgrep: Molecular search program using SMARTS pattern
* obprop: Print standard molecular properties
* obfit: Superimpose two molecules based on a pattern
* obrotamer: Generate conformer/rotamer coordinates
* obchiral: Print molecular chirality information
* obrotate: Rotate dihedral angle of molecules in batch mode
* babel: Convert between various chemical file formats
* obenergy: Calculate the energy for a molecule
* obminimize: Optimize the geometry, minimize the energy for a molecule
* obgrep: Molecular search program using SMARTS pattern
* obprop: Print standard molecular properties
* obfit: Superimpose two molecules based on a pattern
* obrotamer: Generate conformer/rotamer coordinates
* obchiral: Print molecular chirality information
* obrotate: Rotate dihedral angle of molecules in batch mode
Chemeq (Parser for chemical formula
and equilibria)
Chemeq is a basic standalone filter written in C language, flex and bison. It inputs strings like:
2H2 + O2 —> 2 H2O
then it outputs LaTeX code and messages about the equilibrium of a chemical reaction.
example:~/src$ echo “2H2 + O2 —> 2 H2O” | chemeq -lc
2\,H_{2}\,+\,O_{2}\,\rightarrow\,2\,H_{2}O
OK
Chemeq is a basic standalone filter written in C language, flex and bison. It inputs strings like:
2H2 + O2 —> 2 H2O
then it outputs LaTeX code and messages about the equilibrium of a chemical reaction.
example:~/src$ echo “2H2 + O2 —> 2 H2O” | chemeq -lc
2\,H_{2}\,+\,O_{2}\,\rightarrow\,2\,H_{2}O
OK
Easychem (Draw high-quality
molecules and 2D chemical formulas)
EasyChem is a program that helps you creating high quality diagrams of molecules and 2D chemical formulas that can be exported to PDF, PS, LaTeX and fig.
EasyChem was originally developed to create diagrams for chemistry books and is now frequently used for this purpose in commercial and non-commercial chemistry-related books.
EasyChem is a program that helps you creating high quality diagrams of molecules and 2D chemical formulas that can be exported to PDF, PS, LaTeX and fig.
EasyChem was originally developed to create diagrams for chemistry books and is now frequently used for this purpose in commercial and non-commercial chemistry-related books.
Bkchem (Python based chemical
structures editor)
BKchem is a free chemical drawing program, which is written in Python.
Some of the features, you can expect:
* Drawing (bond-by-bond drawing; templates for common rings; expanding of common-groups; draws radicals, charges, arrows; color support …)
* Editing (unlimited undo and redo capabilities; aligning; scaling; rotation (2D, 3D) …)
* Export/Import (fully supported SVG-, OpenOffice.org-Draw-, EPS-export; basic support for CML1 and CML2 import and export)
BKchem is a free chemical drawing program, which is written in Python.
Some of the features, you can expect:
* Drawing (bond-by-bond drawing; templates for common rings; expanding of common-groups; draws radicals, charges, arrows; color support …)
* Editing (unlimited undo and redo capabilities; aligning; scaling; rotation (2D, 3D) …)
* Export/Import (fully supported SVG-, OpenOffice.org-Draw-, EPS-export; basic support for CML1 and CML2 import and export)
Autodock (analysis of ligand binding
to protein structure)
AutoDock is a prime representative of the programs addressing the simulation of the docking of fairly small chemical ligands to rather big protein receptors. Earlier versions had all flexibility in the ligands while the protein was kept rather ridgid. This latest version 4 also allows for a flexibility of selected sidechains of surface residues, i.e., takes the rotamers into account.
The AutoDock program performs the docking of the ligand to a set of grids describing the target protein. AutoGrid pre-calculates these grids.
AutoDock is a prime representative of the programs addressing the simulation of the docking of fairly small chemical ligands to rather big protein receptors. Earlier versions had all flexibility in the ligands while the protein was kept rather ridgid. This latest version 4 also allows for a flexibility of selected sidechains of surface residues, i.e., takes the rotamers into account.
The AutoDock program performs the docking of the ligand to a set of grids describing the target protein. AutoGrid pre-calculates these grids.
Gperiodic (periodic table
application)
GPeriodic is a small X/GTK+-based program which allows you to browse through a periodic table of chemical elements, and view somewhat detailed information on each of the elements. 118 elements are currently listed.
GPeriodic is a small X/GTK+-based program which allows you to browse through a periodic table of chemical elements, and view somewhat detailed information on each of the elements. 118 elements are currently listed.
gElemental (Periodic Table viewer)
gElemental is a GTK+ periodic table viewer that provides detailed information about chemical elements.
It features a table view which allows the elements to be coloured thematically by several properties, a sortable list view and an element properties dialog, displaying a variety of information, including historical, thermodynamic, electrochemical, and crystallographic properties.
gElemental is a GTK+ periodic table viewer that provides detailed information about chemical elements.
It features a table view which allows the elements to be coloured thematically by several properties, a sortable list view and an element properties dialog, displaying a variety of information, including historical, thermodynamic, electrochemical, and crystallographic properties.
xmakemol (A program for visualizing
atomic and molecular systems)
XMakemol is a mouse-based program, written using the LessTif widget set, for viewing and manipulating atomic and other chemical systems. It reads XYZ input and renders atoms, bonds and hydrogen bonds.
Features include:
- Animating multiple frame files
- Interactive measurement of bond lengths, bond angles and torsion angles
- Control over atom/bond sizes
- Exporting to Xpm, Encapsulated PostScript and XYZ formats
- Toggling the visibility of groups of atoms
- Editing the positions of subsets of atoms
XMakemol is a mouse-based program, written using the LessTif widget set, for viewing and manipulating atomic and other chemical systems. It reads XYZ input and renders atoms, bonds and hydrogen bonds.
Features include:
- Animating multiple frame files
- Interactive measurement of bond lengths, bond angles and torsion angles
- Control over atom/bond sizes
- Exporting to Xpm, Encapsulated PostScript and XYZ formats
- Toggling the visibility of groups of atoms
- Editing the positions of subsets of atoms
horae (interactive graphical
processing and analysis of EXAFS data)
ATHENA is an interactive graphical utility for processing EXAFS data. It handles most of the common data handling chores of interest, including deglitching, aligning, merging, background removal, and Fourier transforms.
ARTEMIS is an interactive graphical utility for fitting EXAFS data using theoretical standards from FEFF and sophisticated data modelling along with flexible data visualization and statistical analysis.
HEPHAESTUS is a souped up periodic table for the x-ray absorption spectroscopist. It provides a number of utilities involving tables of absorption coefficients and other chemical data.
ATHENA is an interactive graphical utility for processing EXAFS data. It handles most of the common data handling chores of interest, including deglitching, aligning, merging, background removal, and Fourier transforms.
ARTEMIS is an interactive graphical utility for fitting EXAFS data using theoretical standards from FEFF and sophisticated data modelling along with flexible data visualization and statistical analysis.
HEPHAESTUS is a souped up periodic table for the x-ray absorption spectroscopist. It provides a number of utilities involving tables of absorption coefficients and other chemical data.
PSI3 (Quantum Chemical Program Suite)
PSI3 is an ab-inito quantum chemistry program. It is especially designed to compute properties of small to medium molecules using high-level ab initio techniques.
It can compute energies and gradients for the following methods:
* Closed shell and general restricted open shell Hartree-Fock (RHF/ROHF)
* Complete active space SCF (CASSCF)
* Coupled-cluster singles doubles (CCSD)
Additionally, it can compute energies for the following methods:
* Unrestricted open shell Hartree-Fock (UHF)
* Closed/open shell Moeller-Plesset pertubation theory (MP2)
* Closed shell linear R12 Moeller-Plesset pertubation theory (MP2-R12)
* Configuration-interaction (CI)
* Multireference configuration-interaction (MRCI)
* Coupled-cluster singles doubles with pertubative triples (CCSD(T))
* Closed shell and general restricted open shell equation-of-motion CCSD
It also includes an internal coordinate geometry optimizer.
PSI3 is an ab-inito quantum chemistry program. It is especially designed to compute properties of small to medium molecules using high-level ab initio techniques.
It can compute energies and gradients for the following methods:
* Closed shell and general restricted open shell Hartree-Fock (RHF/ROHF)
* Complete active space SCF (CASSCF)
* Coupled-cluster singles doubles (CCSD)
Additionally, it can compute energies for the following methods:
* Unrestricted open shell Hartree-Fock (UHF)
* Closed/open shell Moeller-Plesset pertubation theory (MP2)
* Closed shell linear R12 Moeller-Plesset pertubation theory (MP2-R12)
* Configuration-interaction (CI)
* Multireference configuration-interaction (MRCI)
* Coupled-cluster singles doubles with pertubative triples (CCSD(T))
* Closed shell and general restricted open shell equation-of-motion CCSD
It also includes an internal coordinate geometry optimizer.
Software
Kimia Gratis
Berikut
adalah beberapa aplikasi yang bisa digunakan untuk mendukung pembelajaran
kimia. Diberikan sedikit paparan dan tautan untuk menuju sumber utamanya
sehingga bisa di-download di komputer. Banyaknya aplikasi visualisasi molekul
ini tentu menjadikan kita punya banyak pilihan disesuaikan dengan kenyamanan
masing-masing pengguna.
- JMol (Platform : Windows, Linux, Mac)
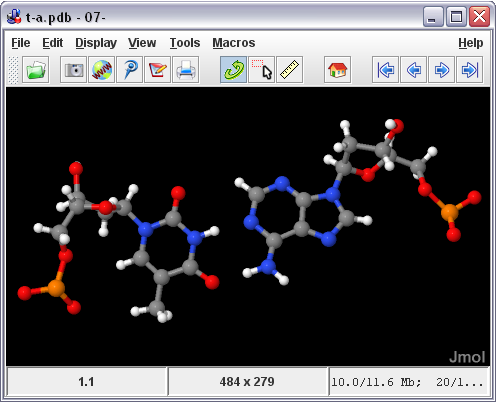 Jmol
ini gartis, merupakan penampil strukutur molekul tiga dimensi (molecule viewer)
yang dapat digukan secara bebas oleh siapapun yang menekuni bidang kimia dan
biokimia. Aplikasi ini merupakan cross-platform, berjalan di sistem operasi
Windows, Mac OS X, dan Linux / Unix. Fitur yang dimilikinya di antaranya
membaca berbagai jenis file dan output dari program kimia kuantum, dan
animasi file multi-frame. JmolApplet adalah applet web browser
yang dapat diintegrasikan ke dalam halaman situs. Aplikasi Jmol adalah aplikasi
Java standalone yang berjalan di desktop. JmolViewer merupakan
seperangkat alat yang dapat diintegrasikan ke dalam aplikasi Java lainnya.
Jmol
ini gartis, merupakan penampil strukutur molekul tiga dimensi (molecule viewer)
yang dapat digukan secara bebas oleh siapapun yang menekuni bidang kimia dan
biokimia. Aplikasi ini merupakan cross-platform, berjalan di sistem operasi
Windows, Mac OS X, dan Linux / Unix. Fitur yang dimilikinya di antaranya
membaca berbagai jenis file dan output dari program kimia kuantum, dan
animasi file multi-frame. JmolApplet adalah applet web browser
yang dapat diintegrasikan ke dalam halaman situs. Aplikasi Jmol adalah aplikasi
Java standalone yang berjalan di desktop. JmolViewer merupakan
seperangkat alat yang dapat diintegrasikan ke dalam aplikasi Java lainnya.
Untuk
menggunakannya silahkan download dan ekstrak file downlad-annya. Klik file Jmol.jar
(di PC mestinya tersedia Javaruntime terlebih dahulu). Buka file yang support
untuk dibuka dengan Jmol.
- ACD/ChemSketch Freeware (Platform : Windows, Linux)
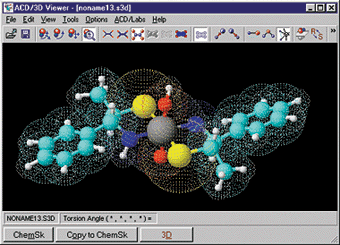 Chemsketch
adalah software grafis untuk menggambar hal yang ada hubungannya dengan kimia .
Bisa menggambar secara manual atau menggunakan templet yang disediakan. Klik
dan gambar molekul, ion, stereobonds, teks, poligon, panah, serta
perlengkapan laboratorium, dll termasuk menentukan secara otomatis massa suatu
atom atau molekul. Kita juga dapat memperkirakan densitas, indeks bias, volume
molar, dll. Selain itu dari ACDLabs juga menawarkan beberapa download
gratis untuk utilitas yang dapat dipergunakan dalam ChemSketch sehingga lebih powerfull.
Chemsketch
adalah software grafis untuk menggambar hal yang ada hubungannya dengan kimia .
Bisa menggambar secara manual atau menggunakan templet yang disediakan. Klik
dan gambar molekul, ion, stereobonds, teks, poligon, panah, serta
perlengkapan laboratorium, dll termasuk menentukan secara otomatis massa suatu
atom atau molekul. Kita juga dapat memperkirakan densitas, indeks bias, volume
molar, dll. Selain itu dari ACDLabs juga menawarkan beberapa download
gratis untuk utilitas yang dapat dipergunakan dalam ChemSketch sehingga lebih powerfull.
Untuk
dapat mendownload software ini diperlukan registrasi (sebentar hanya memasukan
beberapa data alamat email dan lain-lain). Setelelah download lakukan instalasi
sebagaimana biasa dan siap untuk digunakan.
- Molegro Molecular Viewer (Platform: Windows, Linux, dan Mac OS X)

Sumber
http://ahsystemsgroup.com
Aplikasi
gratis dengan multiplatform yang digunakan untuk visualisasi molekul dalam
format PDB, SDF, Mol2, dan MVDML. Fiturnya meliputi pembuatan molekul secara
otomatis, visualisasi permukaan molekul dan backbone.
Ukuran file untuk windows sebesar
5,52 MB
Ukuran file untuk windows sebesar
9,41 MB
Untuk MacOSX saya belum coba,
menungggu untuk punya MacOSX dahulu, bagi yang mau menyumbangkan Mac-nya untuk
saya, dengan senang hati saya menerimanya  .
.
Untuk file-file dengan ekstensi pdb,
bisa dibuat sendiri dengan aplikasi di atas atau men-download-nya dari elchem.kaist.ac.kr. Model molekul yang tersedia di situs ini adalah
molekul-molekul organik dari yang paling sederhana hingga yang cukup kompleks.
- Symyx Chime dan Symyx Draw (Platform: Windows dan MacOSX, Linux belum tersedia)

Sumber
http://blog.goo.ne.jp/
Kedua
aplikasi ini gratis (no fee) siapapun boleh mengunduhnya dan memakainya
secara cuma-cuma. Sebelum mengunduhnya diharuskan melakukan registrasi dengan
mengisikan beberapa data yang empunya situs perlukan. Sayang untuk platform
linux belum tersedia jadi mesti menggunakan Wine agar bisa menjalankannya di
linux. Kedua software ini saling melengkapi sehingga kita bisa memanfaatkannya
dalam program pengajaran di kelas untuk mata pelajaran kimia.
Symyx
Chime versi 2.6 SP8 berformat zip berukuran 3,9 MB. Chime
merupakan plug-in yang secara interaktif menampilkan molekul 2D (dua
dimensi) dan 3D (tiga dimensi) langsung di halaman Web. Kita juga dapat
memutar, memformat, dan menyimpan molekul untuk digunakan dalam program lain.
Symyx
Draw versi 3.3 berukuran cukup besar dalam format zip (66 MB). Dengan aplikasi
ini kita bisa menyisipkan model molekul yang kita buat ke dalam halaman situs,
dokumen, spreadsheet maupun presentasi.
- Kalzium (Platform: Linux)
 Kalzium
adalah nama sebuah software (open source software - OSS) yang memanfaatkan
tabel periodik untuk mengeksplorasi setiap unsur. Tapi ternyata tidak sekedar
tabel periodik biasa, lebih dari sekedar tabel periodik. Kalzium berasal dari
bahasa Jerman dari kata kalsium. Saat ini Kalzium sampai di versi Hidrogen.
Untuk diketahui semenjak Kalzium menjadi aplikasi standalone ia
versinya akan dinamai dengan urutan nama unsur dalam tabel
periodik.Materi-materi pelajaran kimia, baik di kelas 10, kelas 11 IPA, maupun
kelas 12 IPA bab-bab tertentu sangatlah tepat memanfaatkan kehebatan Kalzium.
Tentu ini hanya merupakan alternatif dalam penggunaan media pembelajaran.
Sampai saat ini Kalzium belum mendukung untuk sitem operasi Windows. Hanya bisa
berjalan untuk sistem operasi Linux. Oleh karena itu di sarankan pengguna yang
ingin mencobanya sebaiknya meningstall sistem operasi Linux, gratis dan halal.
Silahkan pilih distro yang disukai.
Kalzium
adalah nama sebuah software (open source software - OSS) yang memanfaatkan
tabel periodik untuk mengeksplorasi setiap unsur. Tapi ternyata tidak sekedar
tabel periodik biasa, lebih dari sekedar tabel periodik. Kalzium berasal dari
bahasa Jerman dari kata kalsium. Saat ini Kalzium sampai di versi Hidrogen.
Untuk diketahui semenjak Kalzium menjadi aplikasi standalone ia
versinya akan dinamai dengan urutan nama unsur dalam tabel
periodik.Materi-materi pelajaran kimia, baik di kelas 10, kelas 11 IPA, maupun
kelas 12 IPA bab-bab tertentu sangatlah tepat memanfaatkan kehebatan Kalzium.
Tentu ini hanya merupakan alternatif dalam penggunaan media pembelajaran.
Sampai saat ini Kalzium belum mendukung untuk sitem operasi Windows. Hanya bisa
berjalan untuk sistem operasi Linux. Oleh karena itu di sarankan pengguna yang
ingin mencobanya sebaiknya meningstall sistem operasi Linux, gratis dan halal.
Silahkan pilih distro yang disukai.- MarvinSketch (Platform: Windows, Linux, MacOS)
 Banyak
hal yang bisa dimanfaatkan dari MarvinSketch untuk pengajaran kimia. Gambar dan
animasi molekul dapat dibuat dengan mudah. Selanjutnya dapat disisipkan dalam
media pembelajaran kimia. Tutorial berupa video untuk mengefektifkan
MarvinSkecth dapat dilihat atau diunduh dari sini. MarvinSketch adalah software kimia hanya untuk visualisasi
rumus struktur kimia yang gratis, bisa dijalankan di sistem operasi Windows dan
Linux juga MacOS. Mau mencicipinya silahkan unduh dari web ChemAxon ini.
Banyak
hal yang bisa dimanfaatkan dari MarvinSketch untuk pengajaran kimia. Gambar dan
animasi molekul dapat dibuat dengan mudah. Selanjutnya dapat disisipkan dalam
media pembelajaran kimia. Tutorial berupa video untuk mengefektifkan
MarvinSkecth dapat dilihat atau diunduh dari sini. MarvinSketch adalah software kimia hanya untuk visualisasi
rumus struktur kimia yang gratis, bisa dijalankan di sistem operasi Windows dan
Linux juga MacOS. Mau mencicipinya silahkan unduh dari web ChemAxon ini.- Ikuti pembaharuan halaman ini untuk melihat berbagai software kimia gratis berikutnya, di mana lagi kalau tidak di halaman ini.

Tidak ada komentar:
Posting Komentar